在基因组学领域,细胞核目前仍是一个未知的领域,我们对于其动态性状了解得如此之少是一件令人尴尬的事。为了解基因组的工作方式,仅知道DNA序列是不够的,我们还必须研究染色体的结构

基因组学的下一个前沿是空间结构,即盘绕在细胞核中的染色体的三维结构。染色体的空间结构并不是随机地将2米长的DNA包装进一个横跨可能10微米的球体中,它是随细胞的类型而变化,并对基因表达产生迄今仍很神秘的影响。德国慕尼黑路德维希马克西米利安大学的遗传学家托马斯·克莱莫(Thomas Cremer)说,破译染色体三维结构对功能的影响将面临很多困难,尤其是研究人员仍在尝试发现染色体是如何随细胞变化而改变的。克莱莫自1970年代以来一直在研究基因组的空间结构,他说:“细胞核仍是一个未知的领域,我们对于其动态性状了解得如此之少是一件多么令人尴尬的事。”
近几十年来已知的基本知识是:DNA双螺旋盘绕着被称为“组蛋白”的蛋白质而形成“染色质”链,染色质链再装配成染色体。但是,当提到染色体自身的扭曲和旋转时,英国剑桥巴布拉汉研究所从事基因组研究的生物学家彼得·弗雷泽(Peter Fraser)认为,其中的远程相互作用似乎难以让人置信。“即便有很多的结构,目前仍不清楚基因组的空间结构在扮演何种角色。人们过去假定,在细胞核中相距5万个碱基的序列不能彼此发现。”

荧光原位杂交能显示出细胞核中基因的位置。图为人类第16号染色体上一个富含基因区域(含50万个碱基)的球形构象
现在,科学家知道这种远程相互作用一直在发生。2002年,弗雷泽的实验室成为了首批检测到“远程环状相互作用”的实验室之一,该作用能使基因序列与远处的调控元件发生物理接触,包括失活的染色质通常被分流到细胞核的边缘(在老鼠的视网膜细胞中这种排列是相反的),能使更多的光到达视网膜的光受体。
基因组的空间结构是非常重要的,如果发生改变,将会带来严重后果。例如,一种被称为“伯基特氏淋巴瘤(Burkitt’s lymphoma)”的淋巴系统癌症,是由第8号染色体中的一段“竖”在第14号染色体上导致的,反之亦然。发生这样的情况,是因为白细胞中的染色体排列自身的方式出了问题――转录期间集合在一起的基因其发生染色体易位的频率更高。已经发现,多种类型的癌症都与影响染色质结构的蛋白质发生突变有关。研究人员推测,不编码基因的DNA片段中发生的与疾病有关的突变,能够改变远程相互作用。
结构中的答案
研究人员很早就已知道,DNA序列和组蛋白被“贴上”了化学修饰的“标签”,这些修饰在“打开”或“关闭”基因的同时,可以给这些“表观遗传”修饰进行分类。新加坡基因组研究所的生物学家阮一俊(音译)认为,染色质的三维结构反映了表观遗传调控的一个更高层次,这一点日益明晰。他开发出一种能发现特异性蛋白质介导的远程相互作用的技术,现正从染色质折叠、移动和通讯的角度寻找答案,而不是假定基因的活性完全是由沿着线性DNA序列的化学附件决定的。而且,这种探讨正逐渐形成包括像“染色质网络”、“染色体相互作用组”和“空间表观遗传学”等概念。

乔布·德克
一系列的技术创新也正开始揭示这些概念的意义。例如,新型的显微镜能让研究人员更准确地看到更多的细胞核,以此开展的实验能允许研究人员确定相互作用的DNA序列,或将DNA序列在细胞核内定位。但挑战依然存在,因为染色体的运动是动态的、不确定的,这给检测带来很大的困难,而搞清楚基因组构造是如何影响基因活性的则更困难。
直到本世纪初,几乎所有对染色体排列的研究都依赖于显微镜技术:研究人员从中能够标记DNA序列或与此相关的分子,然后观测这些标记的区域在细胞核内的位置。但染色质链只有大约10纳米厚,传统荧光显微术的分辨率最高是200纳米,因此能揭示出两个基因座彼此接近,但无法判断它们是否连接在一起。如果相互作用很脆弱或很短暂,显微术也就无能为力了。
英国伍斯特大学医学院从事基因组学研究的生物学家乔布·德克(Job Dekker)早年在哈佛大学的专业方向是研究细胞分裂的力学作用,当时他想绘制出介导染色体间相互作用的DNA序列。一天在去实验室的路上,他突然想到了通过阻碍彼此靠近的两条染色质链来“捕捉”相互作用,从而把双链DNA“溶解”成单一分子。德克说:“刚开始工作就遇到一个难题――两个基因座在三维结构中位于何处?如果通过一系列分子步骤把它转变成一个简单的问题,仅仅是对一个DNA片段进行测序。”
德克的这一想法成就了一项技术,在2002年出版的文献里被称为“染色体构象捕获”,这项技术后来产生了很多变体,但其基本原理是相同的。具体的实验从“交联”开始:用甲醛浸泡细胞,把DNA粘到与之关联的蛋白质上,然后用限制性酶把DNA切碎,或用超声波把DNA剪断,得到的是纠缠在一起的DNA和蛋白质的“毛球”。
进一步的实验方案就不同了,但都要把游离的DNA链结合起来以创建杂交分子――即在同一个毛球上紧密连在一起的DNA链的“结扎”产物。对那些基因(与特定的转录因子或蛋白质有关的基因)感兴趣的研究人员将使用特别设计的抗体来捕获相关的毛球。在一些技术中,化学修饰的核苷酸被整合进杂交分子以利于纯化,而在另一些技术中,正确使用聚合酶链反应(PCR)能扩增感兴趣的基因座附近的DNA序列。
介质的问题
不管使用哪种技术,研究人员在选择限制性酶时都需要仔细。例如,限制性酶在由6个碱基对序列组成的位点切割时,会产生不能捕获相互作用的较大片段;而识别4个碱基对序列的酶能产生更多较小的片段,但或许会制造出太多的背景信息,以致真实的相互作用无法被检测到。
研究人员还需记住,用这种技术生成的大多数杂合DNA分子是随机相互作用的结果,特别是在位于同一个染色体上相距仅几千个碱基的两个基因座之间;把信号从背景噪声中分离出来需要专业的生物信息学知识和重复实验。德克说:“在两年前常常是这种情形,获得数据就意味着到了研究项目的终点。但现在,获得数据意味着项目才开始。”
不同的技术产生了不同的信息。对Hi-C技术(高通量3C技术)而言,100万个已测序分子(或称100万个“读出”)可提供一份完整人的基因组的低分辨图谱;而对4C技术(循环3C)来说,100万个“读出”能为一个感兴趣的基因产生一张详细的相互作用图谱。更进一步说,在“双末端标签测序进行染色质相互作用分析”(ChIA-PET)这一技术中,同样多的数据能表明:哪一些转录因子结合位点与哪一些基因启动子发生了相互作用。
位于加州卡尔斯巴德市的“生命技术”公司的技术开发人员董寿连(音译)说,今年夏天,他们计划开发一种用于与3C实验试剂“捆绑”在一起的试剂盒。该试剂盒允许研究人员监控和优化样品的消化,使用更少的样品用于连接反应,在一天半的时间里就能产生一个连接产物的文库。但是,对通量而言,最重要的因素或许是下一代测序仪日益增长的实用性,如卡尔斯巴德市的“应用生物系统”公司和加州圣地亚哥市的伊鲁米娜公司生产的下一代测序仪,它们能对这些实验中产生的数十万个较短的杂合DNA分子进行快速测序。
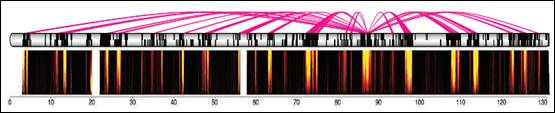
老鼠第8号染色体上同Rad23a基因相互作用的区域,这些相互作用被构象捕捉技术揭示出来
从序列到想法
检测特异性相互作用的基因座的能力已在生物学领域开始显现。去年9月,麻省理工学院的分子生物学家理查德·杨(Richard Young)领导的团队阐述了存在一种生物系统的证据,该系统能将分开的DNA片段拼合在一起,而拼合后的这些片段可控制基因表达。该研究团队发现,在胚胎干细胞中被转录的基因的增强子序列和核心启动子上,常常结合一种“介质”蛋白质复合物。另外一种蛋白质,即能将两个DNA片段连在一起的凝集蛋白(cohesin)与“介质”蛋白质结合在一起,后者可纯化凝集蛋白。在后续的3C研究中表明,在干细胞中启动子和增强子序列之间的相互作用增加了,但在基因不活跃的另一类型的细胞中相互作用没有增加。
荷兰乌特勒支发育生物学研究所的生物学家沃特·德莱特(Wouter de Laat),阐述了如何使用3C技术把一个基因同它的调控因子相匹配的关系。对他来说,染色体捕获技术最令人兴奋的应用是其全面性:它能解决在不同组织中哪些位点与哪些基因相互作用的问题。他说:“有调控可能性的位点比我们现在拥有的基因还多,了解哪个位点对哪个基因起作用的唯一方法就是得到三维结构。那是在功能基因组学中我们需要的下一个层次。”
当前的技术手段还没有强大到足以把调控元件与基因组中的所有基因相匹配,但德莱特和其他的实验室一直在致力于更深远的方法,他们希望能在今年把这些方法发表出来。德莱特说,尽管研究人员倾向于在他们“碰巧”研究的某个基因的环境里解释答案,但这对全基因组提出问题是很有用的。不过由于每个基因都是染色体的一部分,因此,这些研究结果可能小于那些研究同基因“邻居”关系的结果。
还有一个挑战是“从信号到噪声”的问题。当前所有的技术都被用在1000万到2000万个之间的细胞上,这是否意味着所观察到的相互作用代表着平均值?美国国立癌症研究所(NCI)的细胞生物学家汤姆·密斯泰利(Tom Misteli)说:“没有人认为,通过测序技术确定的全部相互作用能发生在任何一个细胞中,其中所发生的任何一个相互作用都将作为一个信号出现,但这种相互作用并不是告诉你它在细胞中发生的频率,这就使解释测序数据变得有点复杂。”
眼见为实
为了找出相互作用发生的频率,研究人员不得不在显微镜下清点标记的细胞的数目。对活细胞成像来说,他们能将表达荧光蛋白的基因插入细胞中,前者可结合到目的DNA位点上,不过这项技术工作量大且冗长乏味。不过,一种固定细胞的技术――荧光原位杂交(FISH)目前用得更多,即先用甲醛对细胞核进行处理,然后令其变性到足以允许用荧光标记了某些序列的DNA探针进入核内。
密斯泰利认为,一般地,通过染色体构象研究识别的相互作用只在显微镜下约十分之一的细胞中能观察到。但这并不意味着相互作用是不真实的,相反,这个比率恰好表明染色体的排列是动态且多变的,同时也说明研究是多么的困难。
去年,弗雷泽和同事联合应用染色体捕获和显微术研究时发现,一个单一的转录因子Klf1可帮助把来自遥远基因座的靶基因带入一个空间中的基因簇中。这种“转录相互作用组学”(transcription interactomics)的研究能揭示细胞分化和稳定性的秘密,但是掌握这些必需的技术是一项艰难的任务。为了从背景信号中分离出相关的杂合分子,研究人员对4C技术做了重大调整,并且为了显示多个基因座能同时集合在一起,团队领导人斯蒂芬·肖恩菲尔德(Stefan Schoenfelder)在显微镜下察看了大约5万个细胞――在暗室里花了半年时间才完成!

汤姆·密斯泰利:“任何相互作用都将作为一个信号出现,但它并没有告诉你它在细胞中发生的频率,这就使它变得复杂。”
这种情形为密斯泰利所熟悉,他于2009年用荧光原位杂交技术揭示了基因在癌症中是如何改变自己位置的。这类知识通常能帮助诊断:当基因活跃时,它们能从细胞核的边缘移向中心,不过个别基因是以不可预测的方式在移动。目前还没有人能全面地察看基因定位,以便发现基因定位在不同细胞类型中可能的变化。密斯泰利说:“当前基因定位研究都基于较少的样品数量和人们特别喜爱的基因,所以你想看更多的基因简直是不可能的。”
技术在不断进步,这让研究人员能看到更多的细胞。弗雷泽说,当前用更快的自动聚焦和更敏捷的机械镜台装备的显微镜,能让肖恩菲尔德在一个月或更短的时间里完成同样数量的实验。包括其他可用的平台,如马萨诸塞州的珀金埃尔默公司销售的一种名为“Opera”的高容量扫描系统,这种设计允许该系统能在要求的高分辨率下工作,以便确定序列在细胞核中的位置。

在基因组中,当给定基因座之间的距离后,基因座之间的相互作用可能比期望的要多或少
“Opera”能在一分钟内检测数百个细胞中的基因座――比独立操作的显微镜快很多――使困难的技术变得更易接近(特别是对非专家而言)。珀金埃尔默公司成像部副总裁阿奇姆·列欧普利茨廷(Achim von Leoprechting)说:“我们正在看到荧光原位杂交逐渐移出了专业化的实验室,因此从成像的观点看,我们需要确保这些实验室能使用这些平台并得到高质量的数据,而不必被训练成显微镜工作者。”
珀金埃尔默公司的一位高容量筛选专家亚伦·李辛格(Aaron Risinger)说,已在研究细胞核内基因位置的研究人员特别渴望能检测不同条件下的更多的细胞类型,“对那些做一次性实验的个人来说,未来的发展就是在高通量上做实验”。事实上,密斯泰利正在这样做,他购买了“Opera”这一研究平台,旨在做超高通量的细胞的生物学成像。
较低通量的技术也有其支持者。伦敦大学帝国理工学院的细胞生物学家安娜·庞波(Ana Pombo)发明了冷冻荧光原位杂交技术:研究人员无需固定和变性完整的细胞,而是把细胞浸泡在一种糖溶液中并进行冷冻,然后把细胞切成薄片再添加DNA探针。这个过程在技术上要求很高,但可比标准的荧光原位杂交产生更少的人工产物和更高的分辨率。因为这些探针不需要穿过完整的细胞核。
电子显微镜有非常高的分辨率,但是细胞的染色和成像要耗费几日。在过去的三年里,研究人员致力于研究超分辨的光学显微镜,这种显微镜使用诸如同步激光脉冲等技术来聚焦活细胞中的15~20纳米的微小结构――传统的光学显微镜分辨极限是200纳米,但这些仪器还没有进入大部分的实验室。
第三种方法
斯德哥尔摩卡罗琳斯卡医学院的表观遗传学家罗尔夫·奥尔森(Rolf Ohlsson)认为,所有的显微术最终是一种“粗糙”的检测技术,测序技术不能表明哪些相互作用是一起发生的,标准荧光显微术不能区别彼此靠近和接触的基因座,甚至超分辨显微镜也无法可靠地做到这点。他说,“在DNA荧光原位杂交和染色体构象捕获之间存在某个地方,尽管它是真实的。”但即便是精确的表示方法也仍将是不够的:确定一个相互作用发生,远比表明一个相互作用影响功能容易。奥尔森问:“你看到的究竟是一个相互作用,还是仅仅的一个碰撞?”
目前有几个团队正尝试使用构象捕获技术来创建计算模型,这些模型可显示不同细胞类型中和处于细胞周期不同阶段的染色体的位置。为了构建模型,研究人员不用实际测量两个基因座间的距离,而是用算法来处理捕获的DNA序列。通过测量观察到基因组的一个区域与另一区域相互作用的频率,并把这个频率同预测的频率进行比较,这些算法程序就能从序列数据中产生“邻近剖面图”。
2009年,德克和同事们构建了人类细胞的一个模型,该模型把30亿个碱基对的基因组分成3000个片段,并绘出了远程相互作用图谱。只是分辨率太低了,不能显示出单个的基因,更不用说预测哪些结合位点可能有助于形成一个特定的构象,不过创建一个更详细的图谱确实是很困难的。构建相互作用图谱需要大约3000万个溶解DNA分子的“读出”,提高分辨率(从1万个碱基对的片段到10万个碱基对的片段)将需要约30亿个读出,因为随着分辨率线性的提高,读出的数量要求呈指数级增长。即使如此,德克和其同事绘制的图谱仍和已确立的关于染色体“领地”的设想是一致的,表明富含基因的区域紧密地混合在一起。
全基因组模型
今年,德克和位于西班牙瓦伦西亚市的费利佩王储研究中心的生物信息学家马克·马蒂-雷诺姆(Marc Marti-Renom)领导的研究团队,发表了在两种不同类型的细胞上完成的3C复写(5C)的结果。他们用数据创建了人类第16号染色体的一个50万个碱基区域的三维模型。该区域包含一组在大多数细胞类型中都很活跃的管家基因,以及另一套仅在一些细胞中活跃的基因。使用相互作用-频率图谱,研究人员为这两种细胞类型构建了染色质模型,这些模型预测了活跃基因聚集于其中的紧缩染色质结构的存在。在两套基因都活跃的细胞中,模型里的染色质折叠成两个“小球”,在只有管家基因活跃的细胞中,只形成一个小球。荧光原位杂交实验证实了在单个细胞中染色质这个区域的形状和比例。
通过从较小的基因组开始,构建在更高分辨率上的全基因组模型也是可能的。去年,费城威斯达研究所长期从事基因表达研究的肯恩·诺玛(Ken-ichi Noma)和同事们采用上述方法,创建了一个分辨率很高的粟酒裂殖酵母全基因组模型。粟酒裂殖酵母只有3条染色体,总共含有大约1400万个碱基对和5000个基因。通过把基因组划分成只有2万个碱基对的众多片段,研究人员计算出了染色质的不同片段彼此靠近的程度,并证实了用显微术获得的几个结果。去年早些时候,由多个实验室组成的一个团队构建了有16条染色体的酿酒酵母基因组的千碱基分辨率模型。
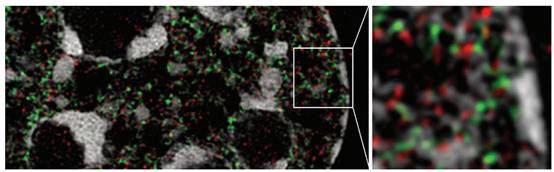
这是老鼠细胞核的部分超分辨图像。图像显示出染色质的密集区域,染色质是通过无DNA通道技术分离得到的。RNA产生和DNA复制发生在这些结构域上的一层解聚染色质中。染色质链偶尔在结构域间很长距离的区域形成环状
目前面临的挑战是从可靠的数据背景中读出其真正的相互作用。华盛顿大学西雅图分校的基因组生物学家威廉·诺布(William Noble)说:“最困难的步骤是,从序列数据里得到一套我们信任且能从功能上解释的相互作用。在论文发表一年前我们手头上就有数据了。”他领导着创建酿酒酵母模型的四个实验室中的一个,进行着结构与人的大脑理解的一个视觉解释研究。但迄今为止,他们仅能获得这种视觉解释的表象。诺布说:“只有在结构绝佳的情况下,我们才引入结构,因为我们不想把任何结论建立在结构本身的基础上。”
其他研究人员承认这类模型可能是有用的,但担心它们可能有误导作用。阮一俊说:“你说两个位点被折叠在一起,那位点之间是什么?我们还没有物理参数来预测那里真正在发生什么。”从高通量数据中得到的与距离估计值之间代表了一个“不现实的平均值”,该平均值没有考虑到染色质是连续的、非定向的和运动的。庞波说:“当你结晶蛋白质时,你能做出它的结构,细胞核可不是那样的。”
模型构建者回应说,未来的表示法将反映出染色体的动态的、半随机的运动,并且通过展示它们的全部趋势,但当前的模型仍是有价值的。德克说:“通过成像,你突出了可变性;通过染色体捕获,你强调了共性。”
克莱莫建议,研究人员在显微镜上花费的时间,应该至少和在电脑上花费的时间一样多。关于高度有序的染色体排列,高通量序列数据能告诉我们什么?人们在真正弄清楚这个问题之前,该领域需要更多的描述性研究。他说:“现在研究人员不得不非常仔细地进行概括,而且我们需要更多的数据。”
资料来源 Nature
责任编辑 则 鸣